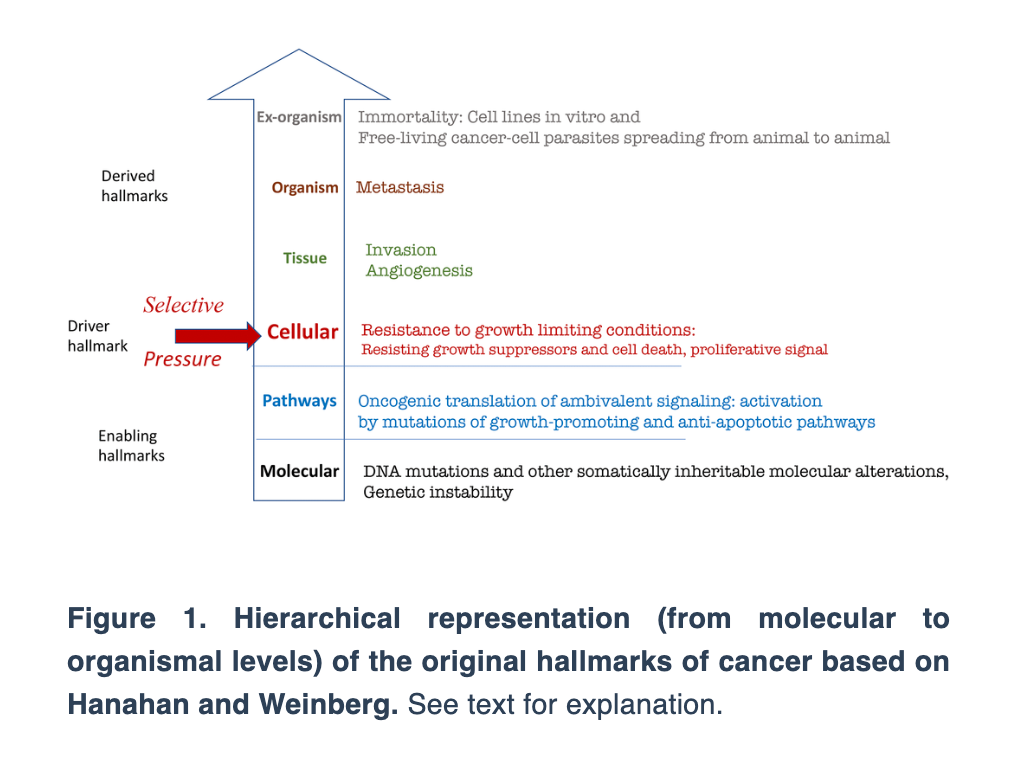
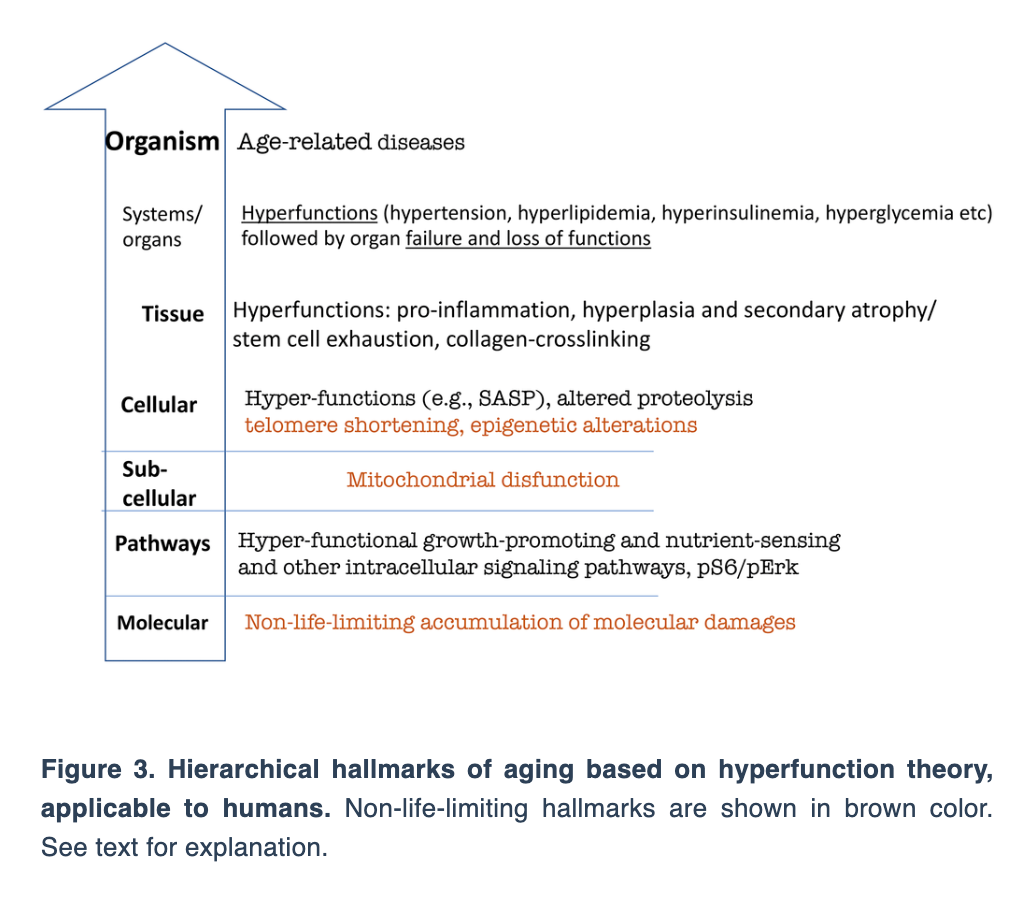
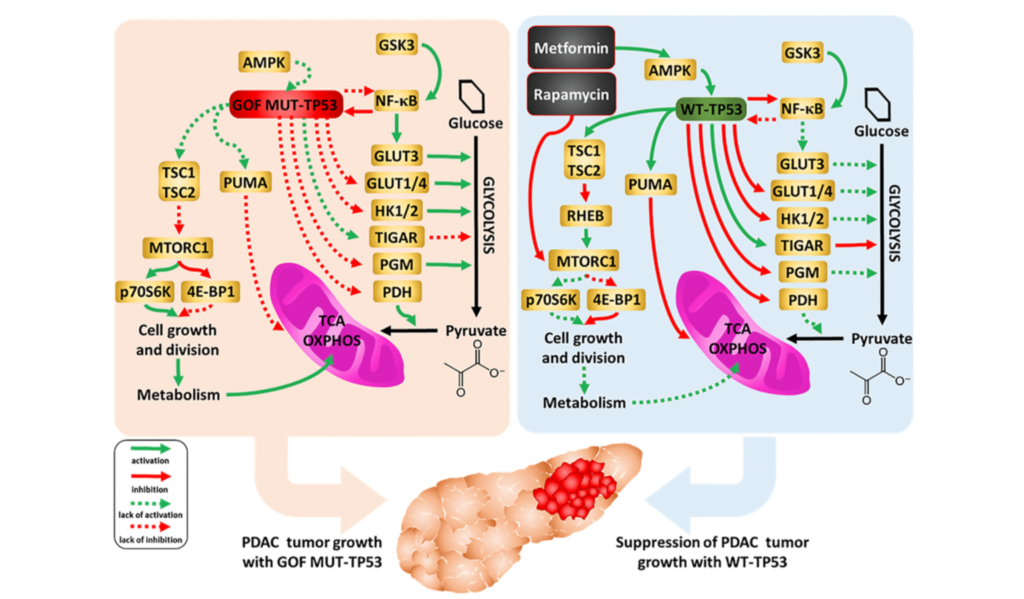
“Here, conceptual similarities between Mikhail Blagosklonny’s hyperfunction theory of aging and Vladimir Dilman’s elevation theory of aging are considered.”
BUFFALO, NY — December 3, 2025 — A new essay was published in Volume 17, Issue 11 of Aging-US on November 19, 2025, titled “On the intergenerational transfer of ideas in aging and cancer research: from the hypothalamus according to V.M. Dilman to the mTOR protein complex according to M.V. Blagosklonny.”
In this work, Aleksei G. Golubev from the N.N. Petrov National Medical Research Center of Oncology reflects on the legacy of two influential Russian scientists, Vladimir M. Dilman and his son Mikhail V. Blagosklonny, who each introduced groundbreaking ideas about aging and cancer. Drawing from his own experience working in Dilman’s lab, Golubev explores how their ideas remain deeply relevant to today’s scientific understanding.
The essay connects Dilman’s “elevation theory” with Blagosklonny’s “hyperfunction theory,” two frameworks that challenge the conventional view of aging as a process of decline. Instead, both propose that aging results from continued biological processes that once supported growth but eventually become harmful when left unchecked.
Dilman believed that aging begins with reduced sensitivity in the hypothalamus, a brain region that regulates the body’s balance. This desensitization disrupts metabolism and hormone levels, setting the stage for many chronic illnesses. Decades later, Blagosklonny expanded on this idea at the molecular level. Central to his theory is the mTOR protein complex, which regulates growth and metabolism and is now a major focus in aging research.
Golubev also explores the historical and personal connections between the two scientists. Dilman, an endocrinologist trained in the Soviet Union, and Blagosklonny, a molecular biologist educated during the post-Soviet period, represent two generations shaped by a shared scientific tradition.
“Dilman’s scientific legacy is not as well recognized as it should be, partly due to bias in citation practices.”
The essay also draws attention to a troubling trend in science: the tendency to overlook early contributions, especially from non-Western scholars. Many of Dilman’s insights, such as the connection between high blood sugar, insulin resistance, and cancer, have since been validated by modern tools, yet his work is rarely cited. Golubev points out how citation practices, language barriers, and historical isolation have contributed to this lack of recognition.
Finally, Golubev encourages the scientific community to look back and acknowledge the foundational work that shaped modern aging science. It also highlights the importance of cross-generational knowledge in moving science forward. By tracing the intellectual journey from hormonal regulation in the brain to molecular pathways in cells, this essay demonstrated the relevance of old ideas in a new biological era.
Paper DOI: https://doi.org/10.18632/aging.206338
Corresponding author: Aleksei G. Golubev – [email protected]
Keywords: aging, gerontology, history of science, hyperfunction, mTOR, hypothalamus, cancer, metabolism, immunity.
Click here to sign up for free Altmetric alerts about this article.
______
To learn more about the journal, please visit www.Aging-US.com and connect with us on social media:
- X
- YouTube
- Bluesky
- Spotify, and available wherever you listen to podcasts
Click here to subscribe to Aging-US publication updates.
For media inquiries, please contact [email protected].