“Pharmacological inhibition of p38 MAPK significantly reduced α-SMA and Col3A1 expression in both TGF-β1-stimulated fibroblasts and primary IPF cells. Mechanistically, TGF-β1-induced expression of α-SMA and Col3A1 was mediated by histone H4K16 acetylation (H4K16ac), which was enriched at gene promoter regions and attenuated by p38 MAPK inhibition.”
Aging has long been linked to a range of biological processes, including cellular senescence, epigenetic changes, and chronic tissue remodeling. Yet, these explanations often describe what happens during aging rather than why certain age-related diseases, such as fibrosis, continue to progress over time. In conditions like idiopathic pulmonary fibrosis (IPF), a key question remains: what drives the persistent activation of cells that should normally return to a resting state after injury? Increasing attention has turned to the interaction between cellular signaling pathways and epigenetic regulation as a potential explanation. Understanding how these processes work together to control gene expression and cell behavior is becoming an important focus in uncovering the mechanisms behind age-related disease.
A new research paper was published in Volume 18 of Aging-US, titled “P38 MAPK is involved in epigenetic regulation of fibrotic genes in replication induced senescence in lung fibroblasts.” The study was led by first author Shan Zhu and corresponding author Yan Y. Sanders from the Department of Biomedical and Translational Sciences, Eastern Virginia Medical School (Macon & Joan Brock Virginia Health Sciences at Old Dominion University), in collaboration with Jennifer Q. Zhou, Kan Wang, and Ming-lei Guo from the same institution.
A Closer Look at Aging, Senescence, and Lung Disease
Aging is often described as a gradual accumulation of cellular damage, but that explanation alone does not fully capture how age-related diseases develop. In conditions like IPF, the problem is not just damage—it is how cells respond to that damage over time. Increasingly, researchers are focusing on cellular senescence, a state in which cells stop dividing but remain metabolically active and can influence their environment in harmful ways.
Understanding how these senescent cells drive disease—and what controls their behavior—has become an important question in aging biology.
Linking Senescence to Fibrosis
IPF is a progressive lung disease strongly associated with aging. One of its defining features is the abnormal activation of fibroblasts, the cells responsible for producing structural components of tissue. When these cells remain activated for too long, they begin to deposit excessive extracellular matrix, leading to scarring and loss of lung function.
In this study, the researchers explored how young (low population doubling level, LPDL) and near-senescent/senescent (high population doubling level, HPDL) lung fibroblasts respond to transforming growth factor-β1 (TGF-β1), a key driver of fibrosis.
Interestingly, both young and senescent cells showed similar increases in fibrotic markers such as α-SMA and Col3A1, suggesting that senescence does not prevent fibroblast activation—but may alter how it is regulated.
A Distinct Role for p38 MAPK Signaling
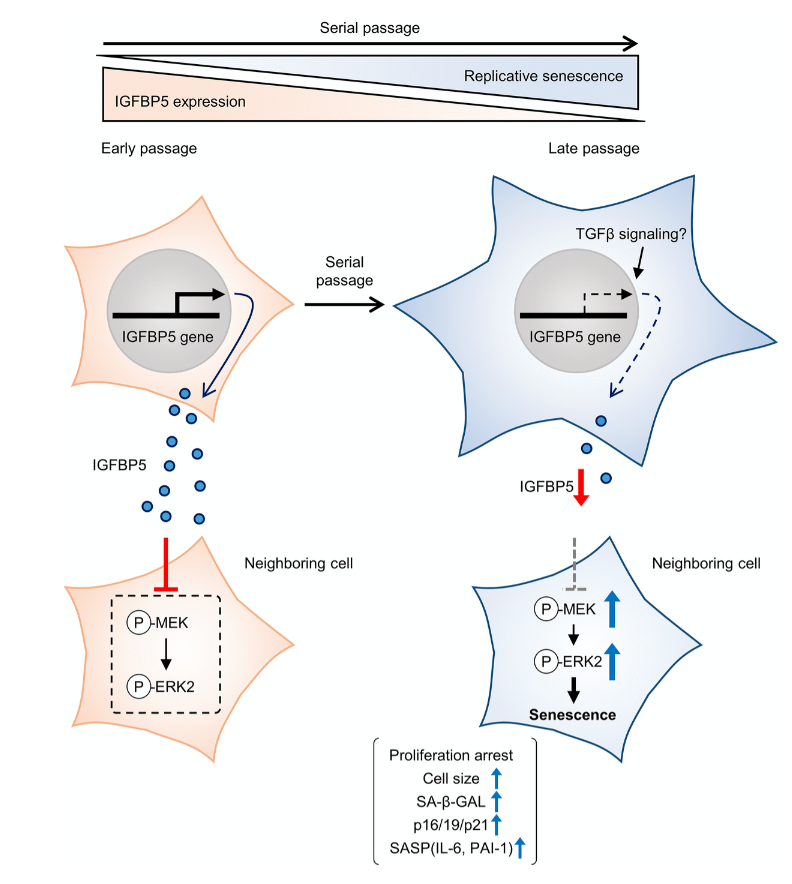
While canonical SMAD signaling behaved similarly in both cell types, the p38 MAPK pathway told a different story. The researchers found a clear difference between the two cell types: p38 MAPK activation was rapid and short-lived in young fibroblasts, but slower and more sustained in senescent cells.
This prolonged signaling in aging cells may help explain why fibrosis becomes persistent and difficult to resolve over time.
Blocking Fibrosis at the Molecular Level
To test whether p38 MAPK plays a functional role, the team used a pharmacological inhibitor (SB202190). The results were clear. Inhibition of p38 MAPK significantly reduced the expression of key fibrotic genes, including α-SMA and Col3A1, and this effect was observed in both experimental fibroblasts and primary IPF patient cells.
These findings suggest that p38 MAPK is not just active during fibrosis but plays an important role in sustaining the fibrotic response.
Epigenetics: The Missing Link
Beyond signaling pathways, the study uncovered an important epigenetic mechanism. The researchers showed that TGF-β1 increases histone H4K16 acetylation (H4K16ac), enriches this modification at fibrotic gene promoters, and that blocking p38 MAPK reduces this effect.
In simple terms, p38 MAPK helps “switch on” fibrosis-related genes by modifying chromatin structure, making them more accessible for transcription.
Why This Matters
Fibrosis is notoriously difficult to treat, in part because it involves multiple overlapping pathways. This study highlights a key intersection between cellular aging (senescence), signal transduction (p38 MAPK), and epigenetic regulation (H4K16ac).
By linking these processes together, the authors provide a more integrated understanding of how fibrosis develops and persists.
Looking Ahead
While this work is preclinical, it points to an important therapeutic opportunity. Targeting p38 MAPK—or the epigenetic mechanisms it controls—could help disrupt the cycle of fibroblast activation and slow disease progression.
Future studies will be needed to explore how these findings translate into clinical settings and whether similar mechanisms operate in other age-related fibrotic diseases.
Conclusion
This study sheds light on how aging-related changes in cell signaling and chromatin structure work together to drive fibrosis. By identifying p38 MAPK as a key regulator of epigenetic activation in fibroblasts, the authors offer a compelling framework for understanding—and potentially targeting—fibrotic disease.
Click here to read the full research paper published in Aging-US.
___
Aging-US is indexed by PubMed/Medline (abbreviated as “Aging (Albany NY)”), PubMed Central, Web of Science: Science Citation Index Expanded (abbreviated as “Aging‐US” and listed in the Cell Biology and Geriatrics & Gerontology categories), Scopus (abbreviated as “Aging” and listed in the Cell Biology and Aging categories), Biological Abstracts, BIOSIS Previews, EMBASE, META (Chan Zuckerberg Initiative) (2018-2022), and Dimensions (Digital Science).
Click here to subscribe to Aging-US publication updates.
For media inquiries, please contact [email protected].